FDA Approves First CRISPR Treatment in U.S.
0 View

- Publish Date:
- 10 December, 2023
- Category:
- Diet
- Video License
- Standard License
- Imported From:
- Youtube
>
It was only 11 years ago that scientists Jennifer Doudna and Emmanuelle Charpentier first described a new way to edit genes, called CRISPR, in a scientific paper. The discovery is so game-changing that the pair earned the Nobel Prize in Chemistry in 2020 for how it could transform the way genetic diseases are treated. Now, on Dec. 8, the U.S. Food and Drug Administration (FDA) approved the very first treatment in the country based on the technology.
[time-brightcove not-tgx=”true”]In the medical world, that’s lightning speed. “It’s incredible,” says Doudna, professor of chemistry and molecular and cell biology at the University of California, Berkeley. “It’s so exciting to see how fast, and frankly how safely and effectively, this therapy is being rolled out in humans.”
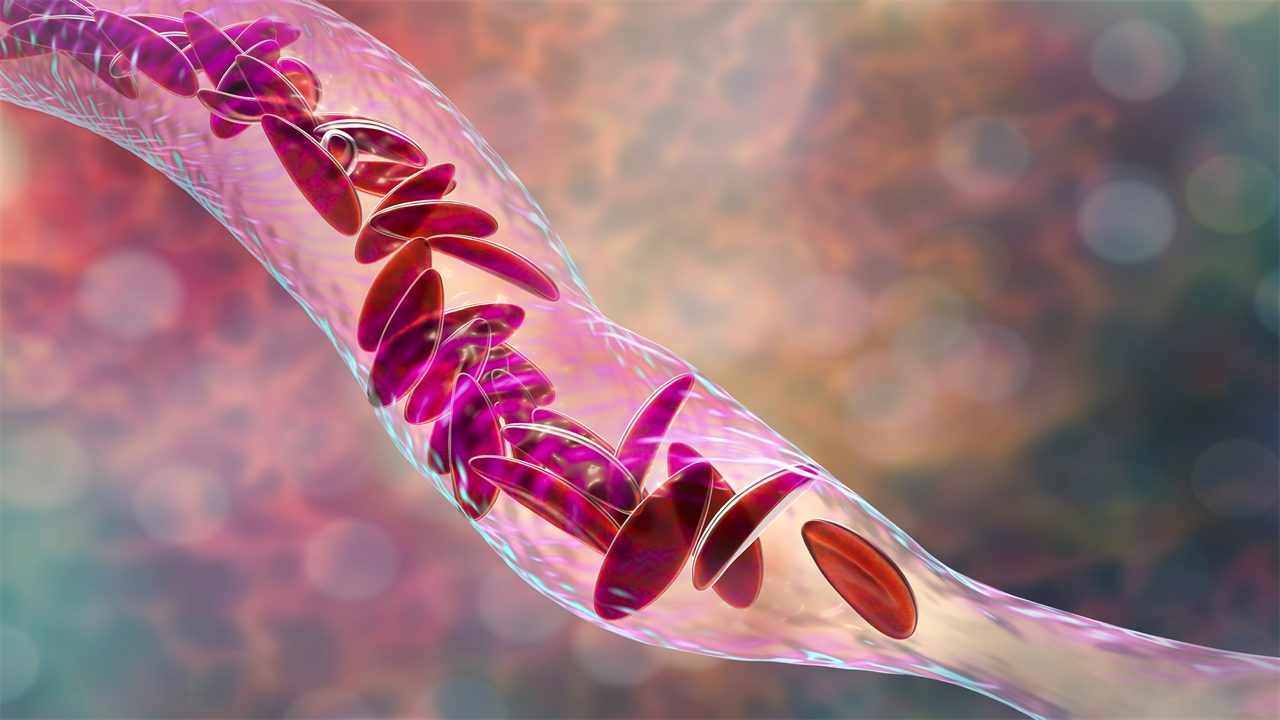
The U.K. had already approved the CRISPR treatment, called exa-cel (brand name: Casgevy), from Vertex Pharmaceuticals and CRISPR Therapeutics (which was co-founded by Charpentier), to treat people with sickle cell disease and beta thalassemia. People with these conditions are born with genetically abnormal blood cells. In the case of sickle cell, the mutations in the gene coding for hemoglobin can cause blood cells to form a sickle shape, rather than a spherical one, and clog up small blood vessels, leading to potentially life-threatening episodes of pain and a higher risk of stroke. Patients with beta thalassemia develop anemia so severe that it can damage organs. Both conditions require lifelong and repeated blood transfusions. CRISPR can increase the population of healthy blood cells in both groups of patients. The FDA approved exa-cel for sickle cell disease, and will make a decision about treating beta thalassemia by March 2024.
The agency also approved another more traditional gene therapy for the disease as well: lova-cel (brand name: Lygenia) from bluebird bio, giving sickle cell patients two powerful new ways of controlling the debilitating and painful attacks that are the hallmark of their disease.
“Gene therapy holds the promise of delivering more targeted and effective treatments, especially for individuals with rare diseases where the current treatment options are limited,” said Dr. Nicole Verdun, director of the office of therapeutic products in the FDA’s Center for Biologics Evaluation and Research, in a statement announcing the approvals.
How exa-cel and lova-cel work
CRISPR is an ideal therapy for these conditions since it involves editing mutated genes in a one-time treatment that could lead to a functional cure. Doctors extract blood stem cells, which produce the body’s entire population of blood and immune cells, from a person’s bone marrow, then grow those cells in the lab. Then, they edit the genes in those cells to boost the production of fetal hemoglobin, which is able to carry more oxygen than adult hemoglobin but normally disappears after birth.
Researchers looked for ways to increase fetal hemoglobin concentration after they learned that about 10% of all people naturally continue to make fetal hemoglobin throughout their adult lives and remain healthy. Among them, people who also had sickle cell disease seemed to have milder forms. Their theory was that increasing the amount of fetal hemoglobin in the blood could help healthy blood cells outcompete the sickled versions and significantly cut down on the chances that the sickled cells would link together and block small vessels. That became the basis of exa-cel.
Exa-cel does this by using CRISPR to target the gene that shuts off fetal hemoglobin. “It’s like removing the stop sign and allowing traffic—in this case, fetal hemoglobin—to go forward down the road,” says Dr. Sharl Azar, medical director of the comprehensive sickle cell disease treatment center at Massachusetts General Hospital.
Lova-cel uses a modified virus that can’t cause disease to introduce a new gene for hemoglobin that mimics the healthy version, with an added anti-sickling feature. Sickled cells tend to form long, stiff chains that can clog vessels and trigger pain, but lova-cel’s hemoglobin “breaks down the chain so they don’t form the long rods anymore,” says Rich Colvin, chief medical officer at bluebird. The end result is that patients have more healthy, unsickled blood cells so they cause fewer painful blockages.
The FDA’s decision
In making its decision on exa-cel, the FDA reviewed a study of 31 patients with sickle cell disease who had experienced repeated blockages in their blood vessels. After getting exa-cel, 29 had no such attacks for a year. While it’s not clear yet how long the effects will last, experts hope that these early results mean longer, and potentially life-long, freedom from hospital visits and painful episodes. For lova-cel, the agency looked at a studyinvolving 32 patients; 28 did not experience any attacks during the two-year study period.
The agency also considered possible side effects. In the case of CRISPR, the most dangerous is off-target editing, in which CRISPR alters the wrong genes or alters genes that can prompt cells to start dividing out of control into a tumor. So far, patients who have received the therapy have not experienced these or other serious side effects. With lova-cel, one of the biggest concerns is where the gene for the healthy hemoglobin is inserted; Colvin says that studies so far show that the gene is inserted up to three times in a single cell, which does not appear to prompt the cell to start dividing abnormally. But “only time will tell whether we have actually done more harm than good by doing these genetic modifications,” says Dr. Markus Mapara, director of the adult bone-marrow transplant and cell-therapy program at Columbia University, who has conducted several gene-therapy trials and has consulted for CRISPR Therapeutics.
one-time treatment, but an arduous roadAs life-changing as both therapies can be, treatment is a grueling, months-long process. Both procedures involve nearly a year of tests and procedures, including an invasive bone marrow transplant. “It’s not for the faint of heart,” says Dr. Monica Bhatia, director of the pediatric stem-cell-transplant program at Columbia University. To be eligible for either therapy, people need to be over 12 years old and have had repeated episodes of blockages due to sickle cell.
The first step is a series of exchange blood transfusions, in which some sickled cells are replaced with healthy cells. The outpatient procedure, which takes place over three to four months, temporarily reduces inflammation and the risk of blockages and attacks leading up to the treatment.
Once the level of sickled cells has dropped low enough, patients are hospitalized so doctors can collect enough stem cells from their bone marrow to be either edited with CRISPR or modified to make healthy hemoglobin, and reinfused back in the patients. Because the bone marrow in sickle cell patients isn’t as robust as that in healthy people, this could take several extraction cycles, and some patients may not even be able to produce enough to qualify for treatment, says Mapara.
If the doctors can recover enough stem cells, they are shipped to Vertex’s and bluebird’s labs, where scientists perform the CRISPR editing, which can take eight to 12 weeks, or the gene therapy so the cells can start producing healthy hemoglobin.
Once the CRISPR or gene therapy cells are ready, patients receive high-dose chemotherapy in order to remove their existing bone marrow and make room for the newly edited cells, which will then seed a population of healthy blood and immune cells. This chemo is likely the most challenging part of the entire procedure—even more difficult, and potentially toxic, than the exa-cel or lova-cel itself. It can also be painful. “I tell my patients that on a scale of zero to 10, this high-dose chemo is close to a nine or 10,” says Bhatia.
Three to four days after chemo, patients finally receive the infusion of CRISPR or gene therapy cells. Then, it’s another four to six weeks in the hospital as doctors monitor them for infections and evaluate how quickly healthy blood cells emerge.
New hope
As involved as the procedures are, they may end up being worth it for most patients, since they could free them from excruciating attacks and even effectively cure their disease. In Bhatia’s patients who have participated in the CRISPR trial, she says that about half of their hemoglobin cells turn healthy and half remain sickled—but that’s enough to keep them out of the hospital and able to attend school and work.
The treatments represent new options for patients who have had very few, says Azar. Hydroxyurea, a drug that is used to treat certain cancers, is the only effective medicine to treat sickle cell; it works by keeping cells rounder rather than sickled, but patients need to continue taking the pills daily to control their pain episodes. Bone marrow transplants to replace sickled cells with healthy ones are also possible, but they’re most effective when the patient and donor are well-matched, making it unavailable for most patients. The best matches come from related donors—but there is only a 25% chance that a sibling would be a match, and because sickle cell is hereditary, patients with the disease have an even smaller chance of having a healthy sibling donor. Transplants from unrelated donors are possible through registries, but their effectiveness is much lower.
“I think this therapy is remarkably transformative,” says Azar of exa-cel. “It gives us options. And it shines a light on a disease that has never had light shone on it before.”
Most sickle cell doctors believe for most patients, both treatments would work equally well in the short term. Having a treatment that could functionally cure sickle cell could also spare patients the long-term organ damage that becomes a legacy of their disease.
The sooner patients are treated, the better chance they have of continuing to live relatively healthy lives with healthy organs, says Mapara. Sickle cell attacks can damage bone, for example, and “once tissue is dead, it’s not reversible,” he says. That’s why he is glad to see that the therapy is approved for anyone 12 years or older, so that even younger patients can take advantage of the opportunity to treat their disease before they develop severe organ damage.